Tutorials
This DeepChem tutorial is designed to serve as a brief primer for antibody design via protein language models. Antibodies are immune proteins also known as immunoglobulins that are naturally produced in the body and bind/inactivate viruses and other pathogens. They are a valuable therapeutic and save lives in immune checkpoint inhibitors for cancer and as neutralizing antibodies for acute viral infection. In addition, they are well known outside the hospital walls for their ability to bind and stick to arbitrary molecular targets, a useful feature in the basic sciences and industrial biochemical facilities.
This tutorial aims to provide a quick overview of key immunology concepts needed to understand antibody structure and function in the broader context of the immune system. We make some assumptions with familiarity with large language models. Take a look at our other tuorial . For the sake of brevity we provide links on non-essential topics that point to external sources wherever possible. Follow along to learn more about the immune system, and protein language models for guided Ab design.
Note: This tutorial is loosely based on the 2023 Nature Biotechnology paper titled "Efficient evolution of human antibodies from general protein language models" [1] by Hie et al. We thank the authors for making their methods and data available and accessible.
1. Immunology 101 ¶
1.1 What is the immune system? ¶
Like other body systems such as the digestive system or the cardiovascular system, the immune system is fundamentally a collection of specialized cells that operate together to accomplish a specific homeostatic function. The immune system is responsible for the protection of our body's vast resources (lipids, carbodhydrates, enzymes, proteins), especially against opportunistic threats of the outside world, such as viruses and bacteria. A very simple yet powerful component of the immune system is the skin, which keeps what's out, out and what's in, in. However, what happens when we get a cut/scrape or when we walk past someone who's coughing and accidentally breathe in the droplets? Thankfully, it turns out the immune system has a whole army of highly specialized white blood cells patrolling our blood and lymph nodes, ready to launch a multi-layered response against these and the countless other cases where foreign objects may enter our bodies. Through these white bloods cells, the immune system is able to accomplish its primary function, self-nonself discrimination , the accurate identification of molecules that are sufficiently dissimilar than those derived from a healthy person.
If you would like to learn more about the complex problem of self non-self discrimination, and appreciate the theory behind the immune system's organization and function, we recommend checking out the following works:
- A Theory of Self-Nonself Discrimination [2]
- The common sense of the self-nonself discrimination, 2005 [3]
- Conceptual aspects of self and nonself discrimination, 2011 [4]
- Self-Nonself Discrimination due to Immunological Nonlinearities: the Analysis of a Series of Models by Numerical Methods, 1987 [5]
- A biological context for the self-nonself discrimination and the regulation of effector class by the immune system, 2005 [6]
1.2 Innate vs. Adaptive Immune System ¶
Remarkably, these white blood cells operate independently, adding and subtracting from the local context (tissue microenvironment), orchestrating cohesive responses in a completely decentralized manner. Individual white blood cells interact with their environments through general cytokine and chemokine surface receptors that facilitate gradient sensing of the local environment. This is the primary axis by which immune cells are able to home to sites of infection/wound healing and send pro-inflammatory or anti-inflammatory signals to neighboring cells. In addition, nearly all immune cells require some level of activation by means of receptor:ligand stimulation by non-self signatures. How specific these receptors are helps highlight the delineation between the two arms of the immune system: the innate and adaptive immune systems.
1.2.1 The Innate Immune System ¶
The innate immune system is the body's first line of defense, and encompasses different types of cells (and proteins) that recognize broadly non-self signals known as pathogen-associated or damage-associated molecular patterns ( PAMPs or DAMPs ). The effector cells of the innate immune system are decorated with ~20 different kinds of pattern recognition receptors (PRRs) that recognize broad signals (single stranded DNA, LPS, other signals of pathogen activity). Being the first responders, the innate immune system handles containment, engulfing the offending signals and otherwise blocking it off, and tries to destroy the threat. Lastly, these cells simultaneously sound the alarm, putting the local area into a heightened state of threat detection and infection prevention, such as spiking a fever and initiating swelling to draw in more immune cells.
1.2.2 The Adaptive Immune System ¶
The adaptive immune system on the other hand, is slow to respond and is often brought in by innate cell activation. Instead of broad PRRs, it relies on tens of millions of somatically rearranged and stochastically generated highly specific adaptive immune receptors (AIRs) whose shape complementarity allows them to identify their complementary molecular patterns called epitopes. There are many more differences between the innate and adaptive immune systems, such as the latter's ability to develop a durable memory response, and those are introduced in greater detail here [7] . It is important to note however, that it is through the independent and asynchronous operation of both the innate and adaptive immune system that we see the dynamics [8] of threat detection, message passing, calling for reinforcements, homing of adaptive immune cells, and activation/expansion of these cells, resulting in complete clearance of the pathogen.Another key distinction between the innate and adaptive immune system is the formation of a robust memory response. Amazingly, evolution has steered this system to imprint past pathogen exposures so that upon re-exposure to the same signal, a small pool of memory cells is activated and antigen-specifc adaptive immune cells rapidly proliferatre and clear the antigen-source [9] .
algorithmic
Adaptive Immune Algorithm
1. Nascent progenitor cells undergoes somatic recombination to stochastically generate an Adaptive Immune Receptor (AIR).
2. Self-selecting methods of ensuring self-tolerance remove cells that react too strongly to the self before being released into the blood.
3. In periphery, naive (antigen-inexperienced) cells interact with antigens via AIRs in search of their cognate epitope.
4. Upon recognition of a sufficiently strong epitope reaction, they divide rapidly and overwhelm the offending antigen with sheer numbers and specialized effector function.
5. After the threat has cleared, this expanded population contracts as cells die without activation signalling, and a lasting pool of memory cells remains.
6. Memory pool persists and is reactivated in an effector state upon antigen reintroduction.
1.2.3 Effector Cells of the Adaptive Immune System ¶
There are two major types of effector cells in the adaptive immune response: T-cells and B-cells . Both the T-cell and B-cell populations in the body are contextualized through their adaptive immune receptors, T-cell Receptors (TCRs) and B-cell Receptors (BCRs), respectively. We can think of both populations as repertoires of receptors, conferring protection against the threats recognized by the repertoire. T-cells help maitnain cellular immunity by interrogating the intracellular component of our bodies' by using their TCRs to scan recycled protein fragments presented at the cell surface (Read more about T-cell mediated immunity here [10] ). B-cells, on the other hand, use their BCRs to survey the extracellular compartment and are tasked with upkeeping humoral immunity : neutralizing threats floating around in the blood and plasma. As effector cells of the adaptive immune response, both T-cell and B-cells are similar in their development and their operation as a unit, though the specifics of per cell function are quite different.
Note : A helpful distinction between antigens and epitopes is that an antigen is something that broadly generates an immune response and can have multiple epitopes. Epitopes are specific molecular patterns that have a matching paratope (binding surface of an adaptive immune receptor).
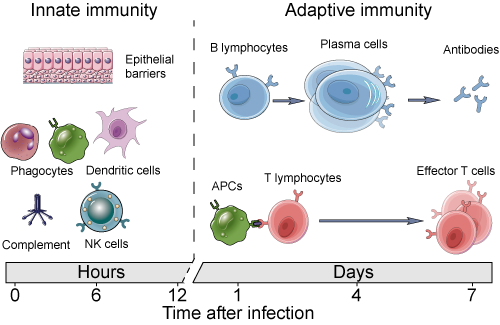
Image Source: Creative Diagnostics
1.3 B-Cells and Antibodies ¶
B-cells, or B-lymphocytes, get their name not from their origin in the bone marrow, but from their discovery in a particular organ of the chicken [11] . These cells circulate in the bloodstream, equipped with unique B-cell receptors (BCRs) that allow them to recognize specific antigens, leading to their activation. This process often requires additional stimulation from helper T-cells which provide essential co-stimulatory signals, an additional layer of verification for non-selfness.
Over the course of the COVID pandemic, whether we wanted to or not were exposed to the concept of antibodies and learned of their association with some sort of protective capacity against SARS-COV-2. But what are they, and where do they originate from?
Antibodies (Abs) are typically represented as Y-shaped proteins that bind to their cognate epitope surfaces with high specificity and affinity, similar to how TCRs and BCRs bind to their epitopes. This is because antibodies are the soluble form of the B-cell receptor that is secreted into the blood upon B-cell activation in the presence of its cognate antigen. The secretion of large amounts of antibodies is the primary effector function of B-cells. Upon activation, a B-cell will divide, with the daughter cells inheriting the same BCR, and some of these cells will differentiate into plasma cells, which are the Ab factories capable of secreting thousands of Abs/min. This is especially useful upon antigen re-encounter where a large amount of antibodies are released by memory cells which neutralize the pathogen even before we develop the symptoms of infection (this is what most common vaccines are designed to do).
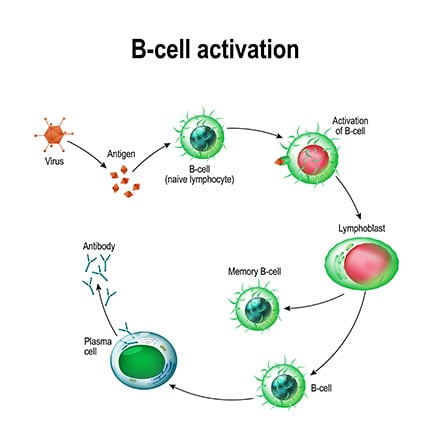
Image Source: Beckman
Neutralizing mechanisms of pathogenesis is only one way that antibody tagging is useful to immune defense. Antibody tagging plays a key role in a number of humoral immunity processes:
-
Neutralization : De-activation of pathogenic function by near complete coating of the functional component of pathogens or toxins by antibodies to inhibit interaction with host cells (i.e. and antibody that binds to the surface glycoproteins on SARS-COV2 now inhibit that virus particle's ability to enter cells expressing ACE2).
-
Opsonization : Partial coating of pathogens enhances rates phagocytosis and removal from the blood by cells of the innate immune system.
-
Agglutination/Precipitation ): Since antibodies have 2 arms (each arm of the Y), they can cross-link and form anitbody-antigens chains which can precipitate out of the plasma and increase their chances of being recognized as aberrant and cleared by phagocytes.
-
Complement Activation : The complement system is a collection of inactive proteins and protein precursors are self-amplifying on activation and help with multiple aspects of humoral immunity. Yet another function of antibodies is their role in initiating the complement cascade that ends in the lysis or phagocytosis of pathogens.
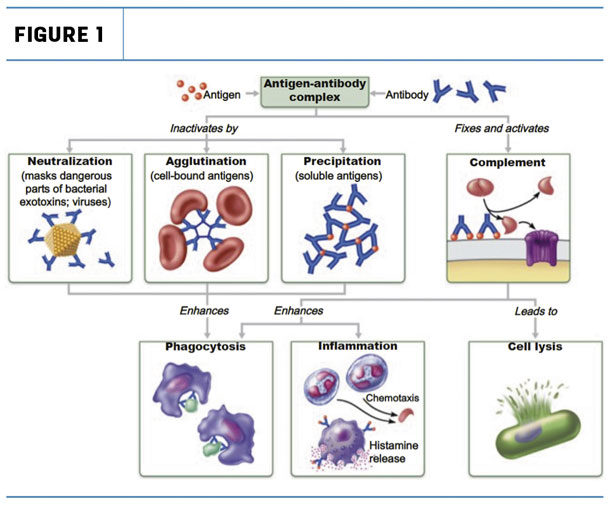
Image Source: The Immune System: Innate and Adaptive Body Defenses Figure 21.15 pulled from [Source]
Given the importance of B-cell mediated immunity, as operationalized by the body's antibody repertoire, it's clear that the diversity of BCR clones plays a critical role in our ability to mount an effective response against a pathogen. The maintenance of a robust BCR repertoire highlights not only the complexity of the immune response but also underscores the potential for leveraging the modularity of this mechanism to introduce new clones for their extraordinary precision in therapeutics such as vaccine development.
1.4 Antibody Sequence, Structure, and Function ¶
The remarkable diversity of antibodies is achieved through somatic recombination, or gene rearrangement at the DNA level that occurs outside of meiosis. The AIR-specific somatic recombination is known as V(D)J recombination and generates both TCR and BCR diversity. During V(D)J recombination, a single gene per set is sampled from the set of variable (V), diversity (D), and joining (J) gene segments and randomly joined together with some baked in error (insertions) to create stable BCRs with unique antigen-binding sites. Additionally, B-cells have an additional process that further amplifies the diversity as well as the functional capacity of antigen-specific antibodies. This process is known as somatic hypermutation. When an activated B-cell divides, somatic hypermutation (SHM) introduces point mutations in the variable regions of BCR generating minor variants of each BCR. These daughter cells compete for survival signals mediated through antigen binding such that only the stronger binders survive.
Structurally, antibodies are composed of two identical light chains and two identical heavy chains, linked by disulfide bonds. Each chain contributes to the formation of the antigen-binding site, located in the variable regions. Within these regions, hypervariable loops known as complementarity determining regions (CDRs) dictate the specificity and affinity of the antibody-antigen interaction. This specificity is measured in terms of affinity using the dissociation constant (Kd), and the avidity (affinity over multi-valent binding sites, see IgM , IgA ). The antibody molecule is divided into two main functional regions:
- Fab Region (Fragment, antigen-binding): Contains the variable regions of the light and heavy chains, responsible for antigen recognition and binding.
- Fc Region (Fragment, crystallizable): Composed of the constant regions of the heavy chains, mediates interactions with innate immune cells and the complement system.
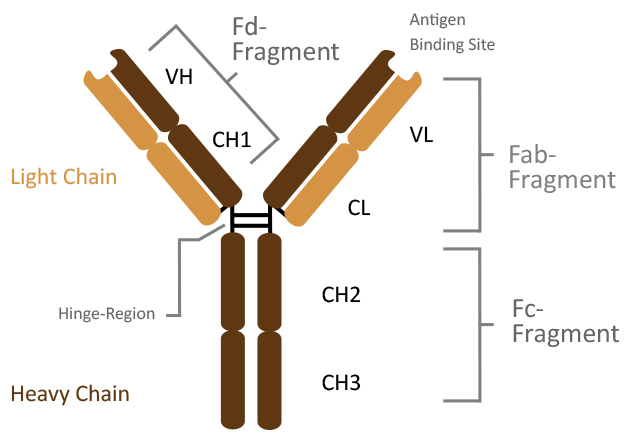
Image Source: Dianova: Antibody Structure \
By harnessing the selection of evolutionary pressures during somatic hypermutation, the B-cell compartment uses a powerful method of further tuning the antibody specificity to have some of the highest affinity interactions in the known protein universe [12] . Their high precision and binding affinities have caused their broad adoption in not only therapeutics but commercial and research applications as well as to tag proteins in solution in flow cytometry, CyTOF, immuno-precipitation, and other target identification assays.
1.5 Current Paradigms for Antibodies as a Therapeutic Modality ¶
Given their unparalleled ability to precisely and durably bind arbitrary targets, there has been a significant interest in possessing antibodies for desired targets. There are a number of therapeutic use cases for these antibodies, for diseases ranging from transplant rejection, non-Hodgkin's lymphoma, immune checkpoint inhibitors for cancer immunotherapy, psoriasis, multiple sclerosis, Crohn's disease, and many more. While antibodies against common pathogens can be isolated from the serum of convalescent individuals and screened for specificity, the process of procuring novel antibodies is substantially more challenging and involves inoculating an animal with an antigen and isolating the antibodies after. For example, anti-venom is a solution of antibodies derived from animals (typically horses) against cytotoxic proteins found in venom. This procedure is both resource and labor intensive. This is because after inoculation and isolation of antibodies from the animal, there is an additional step of screening them for reactivity against the target using reaction chemistry methods such as surface plamon resonance or bio-layer interferometry . As such there has been a great deal of interest in methods of in-silico antibody design. A number of approaches have shown reasonable degrees of success in this task from guided evolution based approaches [1] to newer diffusion based [13] approaches. In this tutorial we will pay homage to the former.
2. Let's Code! Designing Antibodies via Directed Evolution ¶
2.1 Overview ¶
Now that we have the minimal backgrounded needed to understand the antibody design proble and the necessary language model background , we can jump right into antibody design via directed evolution, as shown in the figure below:
\

Image Source: Figure 1. Outeiral et. al
2.2 Setup/Methodology ¶
In Hie et al. the authors decide to use a general protein language model instead of one trained specifically on antibody sequences. They use the ESM-1b and ESM-1v models which were trained on UniRef50 and UniRef90 [14] , respectively. For their directed evolution studies they select seven therapeutic antibodies associated with viral infection spanning Influenza, Ebolavirus, and SARS-COv2. The authors use a straightforward and exhaustive mutation scheduler in mutating every residue in the antigen binding region to every other residue and computing the likelihood of the sequence. Sequences with likelihoods greater than or equal to the WT sequence were kept for experimental validation. For our purposes, we need not be as thorough and can use a slightly expedited method by taking the top-k mutations at a specific point.
Inspired by the work of Hie et al., we first define the pLM driven directed evolution task as simply passing in a masked antibody sequence to a pLM that was previously trained on the masked language modeling objective and examining the token probabilities for the masked amino acids. It really is that easy!
For reference we break the task down into the following steps:
# Antibody Design via pLM Directed Evolution
1. Select a pre-trained model language model (can be pre-trained on all domains or exclusively antibodies)
2. Choose an antibody to mutate.
3. Determine the amino acid(s) to mask out*.
4. Pass the tokenized sequences into the pLM
5. Sample tokens according to a heuristic for increased fitness
*Modification of antibodies needs to focus only on the variable regions as the amino acids at the interface are the ones responsible for driving affinity. Making edits to the constant region would actually be detrimental to antibodies' effector function in the complement system as well as potentially disrupt binding to innate immune receptors. \
2.2.1 Loading the Model + Tokenizer ¶
For this exploration, we pay homage to an early Antibody Language Model AbLang [15] . AbLang is a masked language model based on the RoBERTa [16] model, and pre-trained on antibody sequences from the [observed antibody space (OAS) [17] . AbLang consists of two models, one trained on the heavy chain sequences and one trained on the light chain sequences and the authors demonstrate its usefulness over broader protein language models such as ESM-1b, contradicting the findings put forth in Hie et al. Both the heavy and light chain models are identical in architecture with a $d_{model}$ of 768, max position embedding of 160, and 12 transformer block layers, totaling ~86M parameters.
from transformers import AutoModel, AutoTokenizer, AutoModelForMaskedLM
# Get the tokenizer
tokenizer = AutoTokenizer.from_pretrained('qilowoq/AbLang_light')
# Get the light chain model
mlm_light_chain_model = AutoModelForMaskedLM.from_pretrained('qilowoq/AbLang_light')
# Get the heavy chain model
mlm_heavy_chain_model = AutoModelForMaskedLM.from_pretrained('qilowoq/AbLang_heavy')
/usr/local/lib/python3.10/dist-packages/huggingface_hub/utils/_token.py:89: UserWarning: The secret `HF_TOKEN` does not exist in your Colab secrets. To authenticate with the Hugging Face Hub, create a token in your settings tab (https://huggingface.co/settings/tokens), set it as secret in your Google Colab and restart your session. You will be able to reuse this secret in all of your notebooks. Please note that authentication is recommended but still optional to access public models or datasets. warnings.warn(
tokenizer_config.json: 0%| | 0.00/367 [00:00<?, ?B/s]
vocab.txt: 0%| | 0.00/71.0 [00:00<?, ?B/s]
tokenizer.json: 0%| | 0.00/3.02k [00:00<?, ?B/s]
special_tokens_map.json: 0%| | 0.00/125 [00:00<?, ?B/s]
config.json: 0%| | 0.00/848 [00:00<?, ?B/s]
pytorch_model.bin: 0%| | 0.00/343M [00:00<?, ?B/s]
config.json: 0%| | 0.00/848 [00:00<?, ?B/s]
pytorch_model.bin: 0%| | 0.00/343M [00:00<?, ?B/s]
# Lets take a look at the model parameter count and architecture
n_params = sum(p.numel() for p in mlm_heavy_chain_model.parameters())
print(f'The Ablang model has {n_params} trainable parameters. \n')
mlm_heavy_chain_model
The Ablang model has 85809432 trainable parameters.
RobertaForMaskedLM(
(roberta): RobertaModel(
(embeddings): RobertaEmbeddings(
(word_embeddings): Embedding(24, 768, padding_idx=21)
(position_embeddings): Embedding(160, 768, padding_idx=21)
(token_type_embeddings): Embedding(2, 768)
(LayerNorm): LayerNorm((768,), eps=1e-12, elementwise_affine=True)
(dropout): Dropout(p=0.1, inplace=False)
)
(encoder): RobertaEncoder(
(layer): ModuleList(
(0-11): 12 x RobertaLayer(
(attention): RobertaAttention(
(self): RobertaSelfAttention(
(query): Linear(in_features=768, out_features=768, bias=True)
(key): Linear(in_features=768, out_features=768, bias=True)
(value): Linear(in_features=768, out_features=768, bias=True)
(dropout): Dropout(p=0.1, inplace=False)
)
(output): RobertaSelfOutput(
(dense): Linear(in_features=768, out_features=768, bias=True)
(LayerNorm): LayerNorm((768,), eps=1e-12, elementwise_affine=True)
(dropout): Dropout(p=0.1, inplace=False)
)
)
(intermediate): RobertaIntermediate(
(dense): Linear(in_features=768, out_features=3072, bias=True)
(intermediate_act_fn): GELUActivation()
)
(output): RobertaOutput(
(dense): Linear(in_features=3072, out_features=768, bias=True)
(LayerNorm): LayerNorm((768,), eps=1e-12, elementwise_affine=True)
(dropout): Dropout(p=0.1, inplace=False)
)
)
)
)
)
(lm_head): RobertaLMHead(
(dense): Linear(in_features=768, out_features=768, bias=True)
(layer_norm): LayerNorm((768,), eps=1e-12, elementwise_affine=True)
(decoder): Linear(in_features=768, out_features=24, bias=True)
)
)
2.2.2 Example Antibody Sequence ¶
Next let's choose a heavy chain and light chain for designing. These were chosen from the Ablang examples page on HuggingFace,
# Lets take the variable regions of the heavy and light chains
heavy_chain_example = 'EVQLQESGPGLVKPSETLSLTCTVSGGPINNAYWTWIRQPPGKGLEYLGYVYHTGVTNYNPSLKSRLTITIDTSRKQLSLSLKFVTAADSAVYYCAREWAEDGDFGNAFHVWGQGTMVAVSSASTKGPSVFPLAPSSKSTSGGTAALGCL'
light_chain_example = 'GSELTQDPAVSVALGQTVRITCQGDSLRNYYASWYQQKPRQAPVLVFYGKNNRPSGIPDRFSGSSSGNTASLTISGAQAEDEADYYCNSRDSSSNHLVFGGGTKLTVLSQ'
2.2.3 Masking the Sequence ¶
One of the crucial parameters with this approach is in the determination of which residues to mask and re-design. Let's start off by first setting up some reproducible code so that we can apply the masking procedure to any number of sequences at arbitrary points.
# Sequnece masking convenience function
def mask_seq_pos(sequence: str,
idx: int,
mask='[MASK]'):
'''Given an arbitrary antibody sequence with and a seqeunce index,
convert the residue at that index into the mask token.
'''
cleaned_sequence = sequence.replace(' ', '') # Get ride of extraneous spaces if any
assert abs(idx) < len(sequence), "Zero-indexed value needs to be less than sequence length minus one."
cleaned_sequence = list(cleaned_sequence) # Turn the sequence into a list
cleaned_sequence[idx] = '*' # Mask the sequence at idx
masked_sequence = ' '.join(cleaned_sequence) # Convert list -> seq
masked_sequence = masked_sequence.replace('*', mask)
return masked_sequence
# Test
assert mask_seq_pos('CAT', 1)=='C [MASK] T'
#TODO: Add unit tests with pytest where you can check that the assert has been hit
2.2.3 Model Inference ¶
### Step 1. Mask the light_chain sequence
mask_idx = 9
masked_light_chain = mask_seq_pos(light_chain_example, idx=mask_idx)
### Step 2. Tokenize
tokenized_input = tokenizer(masked_light_chain, return_tensors='pt')
### Step 3. Light Chain Model
mlm_output = mlm_light_chain_model(**tokenized_input)
### Step 4. Decode the outputs to see what the model has placed
decoded_outs = tokenizer.decode(mlm_output.logits.squeeze().argmax(dim=1), skip_special_tokens=True)
print(f'Model predicted: {decoded_outs.replace(" ", "")[9]} at index {mask_idx}')
print(f'Predicted Sequence: {decoded_outs.replace(" ", "")}')
print(f'Starting Sequence: {light_chain_example}')
Model predicted: S at index 9 Predicted Sequence: SADSSSCGVSSTVAHGQTLKINSQGQRHSLYYVRWYQQKPGLAPLLLIYGKNSRPSGIPDRFSGSKSGTTASLTITGLQAEDEADYYCQQSGGSGGHLTVGGGALLATLTQ Starting Sequence: GSELTQDPAVSVALGQTVRITCQGDSLRNYYASWYQQKPRQAPVLVFYGKNNRPSGIPDRFSGSSSGNTASLTISGAQAEDEADYYCNSRDSSSNHLVFGGGTKLTVLSQ
2.2.4 HuggingFace Pipeline Object ¶
Hold on, given the tokenized input with only one masked token, we would expect to see only one change to the the sequence. However, what we get back is something a lot more different that what we put in. Luckily, there's something in the HuggingFace software suite that we can use to address this: Pipelines
HuggingFace Pipelines:
- Pipeline object is a wrapper for inference and can be treated like an object for API calls
- There is a fill-mask pipeline that we can use which accepts a single mask token in out input and outputs a dictionary of the score of that sequence, the imputed token, and the reconstructed full sequence.
Lets see it in action:
from transformers import pipeline
filler = pipeline(task='fill-mask', model=mlm_light_chain_model, tokenizer=tokenizer)
filler(masked_light_chain) # fill in the mask
[{'score': 0.13761496543884277,
'token': 7,
'token_str': 'S',
'sequence': 'G S E L T Q D P A S S V A L G Q T V R I T C Q G D S L R N Y Y A S W Y Q Q K P R Q A P V L V F Y G K N N R P S G I P D R F S G S S S G N T A S L T I S G A Q A E D E A D Y Y C N S R D S S S N H L V F G G G T K L T V L S Q'},
{'score': 0.1152879148721695,
'token': 6,
'token_str': 'E',
'sequence': 'G S E L T Q D P A E S V A L G Q T V R I T C Q G D S L R N Y Y A S W Y Q Q K P R Q A P V L V F Y G K N N R P S G I P D R F S G S S S G N T A S L T I S G A Q A E D E A D Y Y C N S R D S S S N H L V F G G G T K L T V L S Q'},
{'score': 0.0989701896905899,
'token': 9,
'token_str': 'N',
'sequence': 'G S E L T Q D P A N S V A L G Q T V R I T C Q G D S L R N Y Y A S W Y Q Q K P R Q A P V L V F Y G K N N R P S G I P D R F S G S S S G N T A S L T I S G A Q A E D E A D Y Y C N S R D S S S N H L V F G G G T K L T V L S Q'},
{'score': 0.08586061000823975,
'token': 14,
'token_str': 'A',
'sequence': 'G S E L T Q D P A A S V A L G Q T V R I T C Q G D S L R N Y Y A S W Y Q Q K P R Q A P V L V F Y G K N N R P S G I P D R F S G S S S G N T A S L T I S G A Q A E D E A D Y Y C N S R D S S S N H L V F G G G T K L T V L S Q'},
{'score': 0.07652082294225693,
'token': 8,
'token_str': 'T',
'sequence': 'G S E L T Q D P A T S V A L G Q T V R I T C Q G D S L R N Y Y A S W Y Q Q K P R Q A P V L V F Y G K N N R P S G I P D R F S G S S S G N T A S L T I S G A Q A E D E A D Y Y C N S R D S S S N H L V F G G G T K L T V L S Q'}]
Congratulations you have now designed 5 new antibodies!
Disclaimer: For a more thorough antibody (re)design, we will typically want to follow an approach like what was done in Hie et al. where every point along the sequence will be mutated and the total number of sequences will be collated and scored with the top-100 or so antibodies being expressed for validation. If you would like to explore this feel free to try it out yourself as a challenge!
You can also refer to the real data in Hie et al. to see if any of the predicted ones were found to work well and increase fitness.
2.3 Limitations ¶
While promising, this approach is obviously not without its shortcomings. Key limitations include:
- Fixed length antibody design since masked tokens are applied in a 1:1 fashion.
- Lack of target information included during conditional sampling step which can influence choice of amino acid given the sequence context.
- Approach is sensitive to choice of protein language model
This letter [18] provides a great synopsis of Hie et al.'s work, which by extension apply to the methods presented in this tutorial as well.
Citing this Tutorial ¶
If you found this tutorial useful, please consider citing it as:
@manual{Bioinformatics,
title={An Introduction to Antibody Design Using Protein Language Models},
organization={DeepChem},
author={Karthikeyan, Dhuvarakesh and Menezes, Aaron},
howpublished = {\url{https://github.com/deepchem/deepchem/blob/master/examples/tutorials/DeepChem_AntibodyTutorial_Simplified.ipynb}},
year={2024},
}
Works Cited ¶
[1] Hie, B.L., Shanker, V.R., Xu, D. et al. Efficient evolution of human antibodies from general protein language models. Nat Biotechnol 42, 275–283 (2024). https://doi.org/10.1038/s41587-023-01763-2
[2] Bretscher, P., & Cohn, M. (1970). A Theory of Self-Nonself Discrimination. Science, 169(3950), 1042–1049. doi:10.1126/science.169.3950.1042
[3] Cohn, M. The common sense of the self-nonself discrimination. Springer Semin Immun 27, 3–17 (2005). https://doi.org/10.1007/s00281-005-0199-1
[4] Gonzalez S, González-Rodríguez AP, Suárez-Álvarez B, López-Soto A, Huergo-Zapico L, Lopez-Larrea C. Conceptual aspects of self and nonself discrimination. Self Nonself. 2011 Jan;2(1):19-25. doi: 10.4161/self.2.1.15094. Epub 2011 Jan 1. PMID: 21776331; PMCID: PMC3136900.
[5] ROB J. DE BOER, PAULINE HOGEWEG, Self-Nonself Discrimination due to Immunological Nonlinearities: the Analysis of a Series of Models by Numerical Methods, Mathematical Medicine and Biology: A Journal of the IMA, Volume 4, Issue 1, 1987, Pages 1–32, https://doi.org/10.1093/imammb/4.1.1
[6] Cohn, M. A biological context for the self-nonself discrimination and the regulation of effector class by the immune system. Immunol Res 31, 133–150 (2005). https://doi.org/10.1385/IR:31:2:133
[7] Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001. Principles of innate and adaptive immunity. Available from: https://www.ncbi.nlm.nih.gov/books/NBK27090/
[8] Perelson, A. Modelling viral and immune system dynamics. Nat Rev Immunol. 2. , 28–36 (2002). https://doi.org/10.1038/nri700
[9] Shittu, A. (n.d.). Understanding immunological memory. ASM.org. https://asm.org/articles/2023/may/understanding-immunological-memory
[10] Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001. Chapter 8, T Cell-Mediated Immunity. Available from: https://www.ncbi.nlm.nih.gov/books/NBK10762/
[11] Glick, B., Chang, T. S., & Jaap, R. G. (1956). The Bursa of Fabricius and Antibody Production. Poultry Science, 35(1), 224–225. doi:10.3382/ps.0350224
[12] Nooren, I. M. (2003). NEW EMBO MEMBER’S REVIEW: Diversity of protein-protein interactions. EMBO Journal, 22(14), 3486–3492. https://doi.org/10.1093/emboj/cdg359
[13] Karolis Martinkus, Jan Ludwiczak, Kyunghyun Cho, Wei-Ching Liang, Julien Lafrance-Vanasse, Isidro Hotzel, Arvind Rajpal, Yan Wu, Richard Bonneau, Vladimir Gligorijevic, & Andreas Loukas. (2024). AbDiffuser: Full-Atom Generation of in vitro Functioning Antibodies.
[14] Baris E. Suzek, Hongzhan Huang, Peter McGarvey, Raja Mazumder, Cathy H. Wu, UniRef: comprehensive and non-redundant UniProt reference clusters, Bioinformatics, Volume 23, Issue 10, May 2007, Pages 1282–1288, https://doi.org/10.1093/bioinformatics/btm098
[15] Tobias H Olsen, Iain H Moal, Charlotte M Deane, AbLang: an antibody language model for completing antibody sequences, Bioinformatics Advances, Volume 2, Issue 1, 2022, vbac046, https://doi.org/10.1093/bioadv/vbac046
[16] Yinhan Liu, Myle Ott, Naman Goyal, Jingfei Du, Mandar Joshi, Danqi Chen, Omer Levy, Mike Lewis, Luke Zettlemoyer, & Veselin Stoyanov. (2019). RoBERTa: A Robustly Optimized BERT Pretraining Approach.
[17] Olsen TH, Boyles F, Deane CM. Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. 2022 Jan;31(1):141-146. doi: 10.1002/pro.4205. Epub 2021 Oct 29. PMID: 34655133; PMCID: PMC8740823.
[18] Outeiral, C., Deane, C.M. Perfecting antibodies with language models. Nat Biotechnol 42, 185–186 (2024). https://doi.org/10.1038/s41587-023-01991-6